These are the posts of the old forum. It was not possible to transfer the user data, so they are missing in most of the posts. For new questions, go to the general discussions.
Non-specific binding/data modeling issue
- OldForum
- Topic Author
- New Member
-

Less
More
- Thank you received: 0
14 years 2 months ago - 12 years 6 months ago #1
by OldForum
Non-specific binding/data modeling issue was created by OldForum
Hi all,
Thanks for keeping this forum up--as a newer SPR user, it's really been helpful to see folks who've had similar problems and how to get around them.
I'm trying to characterize a novel protein-protein interaction between an unstructured ~80 kDa ligand and a globular, ~40 kDa analyte using a Reichert SR7500 instrument.We've hypothesized that the interaction depends in part upon the C-terminal region of the ligand, so I've tried using two ligand proteins: a full-length one (with the binding region intact), and a slightly-truncated one (with that region removed).
The running buffer I'm using contains 10 mM HEPES, pH 7.4, 2 mM MgCl2, 1mM EGTA, 0.005% TWEEN, 0.1 mg/mL BSA (to cut down on nonspecific protein-protein binding--see below) and varying amounts of NaCl (150 mM and up). The ligand is coupled to a carboxymethyldextran chip, using amine coupling. After some early attempts at steady-state measurements at low flow rates, I've switched to higher flow rates (~50 uL/min) per the manufacturer's suggestion, and have mostly done kinetic titrations due to the difficulty of regenerating the chip (requires 6 M GuCl and a few hours of buffer flow to re-equilibrate).
I've had excellent coupling of ligand to the chip (using just 10mM HEPES pH 8 and some salt--wouldn't want BSA on the chip), and have done most of my measurements with 500-1000 RU of ligand attached. My first round or two of experiments (with no BSA in the buffers) gave me significant binding of the analyte to the ligand, with (at least) two weird features.
1) I couldn't seem to saturate the binding capacity of the chip. By my back of the envelope math, I figured that ~500 RU of bound ligand should lead to ~250 RU of bound analyte if every ligand protein were a) active and b) bound one analyte protein (based on the ratio of their molecular weights). However, I've gotten substantially more binding than that, even at tens-of-micromolar concentrations of analyte.
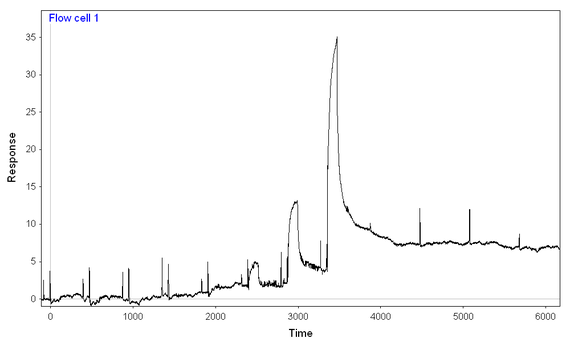
2) At lower concentrations, the reaction seems to reach a "steady-state" level, while at higher concentrations, the binding just keeps on coming (see picture)
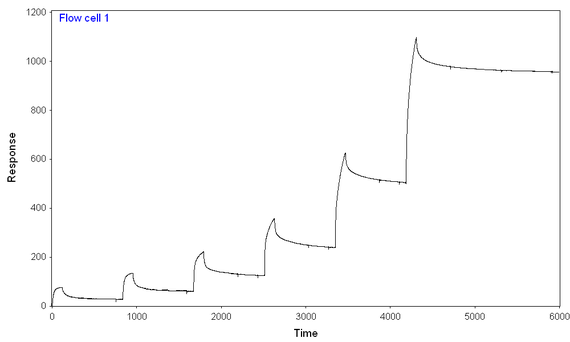
The dissociation in the reaction seems particularly slow, and it almost seems to have two components--a rapid one and an incredibly slow one (that still doesn't get close to baseline, even if left running overnight).
Given these results, I was concerned about nonspecific protein-protein binding, leading me to add BSA to the buffers and increase the salt. Blanks containing BSA show no more binding of BSA to the activated+liganded+inactivated channel than a control activated-and-inactivated one.
Unfortunately, adding BSA and increasing the salt (stepwise to 300 mM and 500 mM) doesn't seem to have fixed the problem. The new data looks clean, but hasn't addressed the nonsaturability or the two-component nature of the binding.
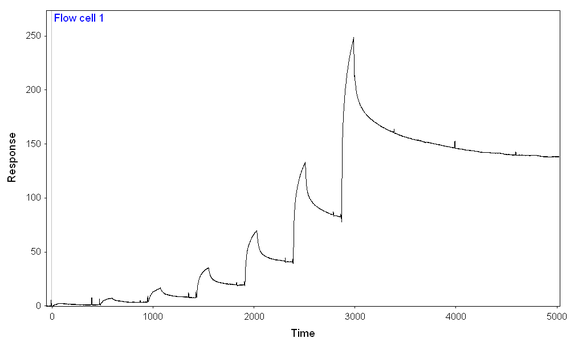
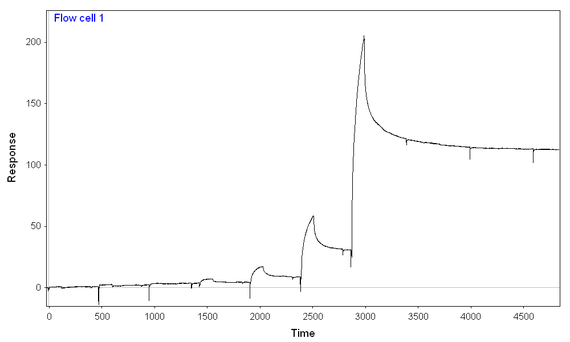
At 300 mM NaCl and 0.1 mg/mL BSA, I performed a 7-step kinetic titration (using the same concentrations of analyte over chips with full-length or truncated analyte), and I still have binding of analyte to both ligands, full-length and truncated. The amount of full length analyte immobilized is ~2/3 of the amount of truncated analyte immobilized. I think I'm seeing more binding of analyte to the full-length ligand than truncated ligand, given the fact that the signal starts at lower analyte concentrations and gets to higher peaks at every given analyte concentration) especially given the difference in immobilization.
Full-length:
Truncated:
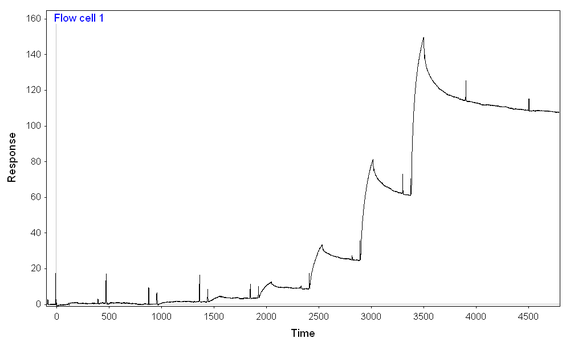
At 500 mM NaCl and 0.1 mg/mL BSA (and an 8-step kinetic titration, again using the same concentrations of analyte for both chips and with the same difference in immobilization), I get more binding to the full-length ligand than to the truncated one (earlier onset for full-length ligand and much higher signal), but still seem to have two-components to the dissocation and difficult-to-model binding.
Full-length:
Truncated:
If anyone has suggestions on how to a) alter the experiment to get nice clean data (I really want a Kd!), b) have alternate approaches to the data analysis (I'd even settle for a "full-length ligand binds analyte x-fold more than truncated ligand does") or c) help me with my issues with CLAMP (which a--only lets me program 6 injections when I need 7-8 and b--seems to shut down every time I ask it to fit the data), I'd really appreciate it.
Thanks and have a great weekend.
Thanks for keeping this forum up--as a newer SPR user, it's really been helpful to see folks who've had similar problems and how to get around them.
I'm trying to characterize a novel protein-protein interaction between an unstructured ~80 kDa ligand and a globular, ~40 kDa analyte using a Reichert SR7500 instrument.We've hypothesized that the interaction depends in part upon the C-terminal region of the ligand, so I've tried using two ligand proteins: a full-length one (with the binding region intact), and a slightly-truncated one (with that region removed).
The running buffer I'm using contains 10 mM HEPES, pH 7.4, 2 mM MgCl2, 1mM EGTA, 0.005% TWEEN, 0.1 mg/mL BSA (to cut down on nonspecific protein-protein binding--see below) and varying amounts of NaCl (150 mM and up). The ligand is coupled to a carboxymethyldextran chip, using amine coupling. After some early attempts at steady-state measurements at low flow rates, I've switched to higher flow rates (~50 uL/min) per the manufacturer's suggestion, and have mostly done kinetic titrations due to the difficulty of regenerating the chip (requires 6 M GuCl and a few hours of buffer flow to re-equilibrate).
I've had excellent coupling of ligand to the chip (using just 10mM HEPES pH 8 and some salt--wouldn't want BSA on the chip), and have done most of my measurements with 500-1000 RU of ligand attached. My first round or two of experiments (with no BSA in the buffers) gave me significant binding of the analyte to the ligand, with (at least) two weird features.
1) I couldn't seem to saturate the binding capacity of the chip. By my back of the envelope math, I figured that ~500 RU of bound ligand should lead to ~250 RU of bound analyte if every ligand protein were a) active and b) bound one analyte protein (based on the ratio of their molecular weights). However, I've gotten substantially more binding than that, even at tens-of-micromolar concentrations of analyte.
2) At lower concentrations, the reaction seems to reach a "steady-state" level, while at higher concentrations, the binding just keeps on coming (see picture)
The dissociation in the reaction seems particularly slow, and it almost seems to have two components--a rapid one and an incredibly slow one (that still doesn't get close to baseline, even if left running overnight).
Given these results, I was concerned about nonspecific protein-protein binding, leading me to add BSA to the buffers and increase the salt. Blanks containing BSA show no more binding of BSA to the activated+liganded+inactivated channel than a control activated-and-inactivated one.
Unfortunately, adding BSA and increasing the salt (stepwise to 300 mM and 500 mM) doesn't seem to have fixed the problem. The new data looks clean, but hasn't addressed the nonsaturability or the two-component nature of the binding.
At 300 mM NaCl and 0.1 mg/mL BSA, I performed a 7-step kinetic titration (using the same concentrations of analyte over chips with full-length or truncated analyte), and I still have binding of analyte to both ligands, full-length and truncated. The amount of full length analyte immobilized is ~2/3 of the amount of truncated analyte immobilized. I think I'm seeing more binding of analyte to the full-length ligand than truncated ligand, given the fact that the signal starts at lower analyte concentrations and gets to higher peaks at every given analyte concentration) especially given the difference in immobilization.
Full-length:
Truncated:
At 500 mM NaCl and 0.1 mg/mL BSA (and an 8-step kinetic titration, again using the same concentrations of analyte for both chips and with the same difference in immobilization), I get more binding to the full-length ligand than to the truncated one (earlier onset for full-length ligand and much higher signal), but still seem to have two-components to the dissocation and difficult-to-model binding.
Full-length:
Truncated:
If anyone has suggestions on how to a) alter the experiment to get nice clean data (I really want a Kd!), b) have alternate approaches to the data analysis (I'd even settle for a "full-length ligand binds analyte x-fold more than truncated ligand does") or c) help me with my issues with CLAMP (which a--only lets me program 6 injections when I need 7-8 and b--seems to shut down every time I ask it to fit the data), I'd really appreciate it.
Thanks and have a great weekend.
Last edit: 12 years 6 months ago by OldForum.
Please Log in or Create an account to join the conversation.
- OldForum
- Topic Author
- New Member
-
Less
More
- Thank you received: 0
14 years 2 months ago #2
by OldForum
Replied by OldForum on topic Non-specific binding/data modeling issue
Hi JWaits,
First the easy part:
Clamp allows only 6 injections, thus you have to do with this.
For the fitting, try to guess the initial values closely and run a dummy. Fix some parameters so they are not fitting making it for the algorithm easier to fit the others. Then progress with fitting one by one the fixed parameters.
I looked up one publication: Beattie, J. et al. Molecular interactions in the insulin-like growth factor (IGF) axis: a surface plasmon resonance (SPR) based biosensor study. Mol.Cell Biochem. .: (2007).
This can you give some ideas to change your experiments.
Arnoud
First the easy part:
Clamp allows only 6 injections, thus you have to do with this.
For the fitting, try to guess the initial values closely and run a dummy. Fix some parameters so they are not fitting making it for the algorithm easier to fit the others. Then progress with fitting one by one the fixed parameters.
I looked up one publication: Beattie, J. et al. Molecular interactions in the insulin-like growth factor (IGF) axis: a surface plasmon resonance (SPR) based biosensor study. Mol.Cell Biochem. .: (2007).
This can you give some ideas to change your experiments.
Arnoud
Please Log in or Create an account to join the conversation.
- OldForum
- Topic Author
- New Member
-
Less
More
- Thank you received: 0
14 years 2 months ago #3
by OldForum
Replied by OldForum on topic Non-specific binding/data modeling issue
I looked up some other publications which may be of use.
Because every interaction has its own merits there is not one solution to this kind of interaction behavior. I have to admit that sometimes we are unable to solve this kind of problems.
In addition to what you have already done, you can try to reverse the system, try a different immobilization strategy and check your proteins for heterogeneity.
1. McGovern, S. L. et al; A specific mechanism of nonspecific inhibition. J.Med.Chem. (46): 4265-4272; 2003.
2. Ryan, A. J. et al; Effect of detergent on "promiscuous" inhibitors. J.Med.Chem. (46): 3448-3451; 2003.
3. Svitel, J. et al; Combined affinity and rate constant distributions of ligand populations from experimental surface binding kinetics and equilibria. Biophys.J. (84): 4062-4077; 2003.
4. Svitel, J. et al; Probing the functional heterogeneity of surface binding sites by analysis of experimental binding traces and the effect of mass transport limitation. Biophys.J. (92): 1742-1758; 2007.
Let us know what your results are.
Arnoud
Because every interaction has its own merits there is not one solution to this kind of interaction behavior. I have to admit that sometimes we are unable to solve this kind of problems.
In addition to what you have already done, you can try to reverse the system, try a different immobilization strategy and check your proteins for heterogeneity.
1. McGovern, S. L. et al; A specific mechanism of nonspecific inhibition. J.Med.Chem. (46): 4265-4272; 2003.
2. Ryan, A. J. et al; Effect of detergent on "promiscuous" inhibitors. J.Med.Chem. (46): 3448-3451; 2003.
3. Svitel, J. et al; Combined affinity and rate constant distributions of ligand populations from experimental surface binding kinetics and equilibria. Biophys.J. (84): 4062-4077; 2003.
4. Svitel, J. et al; Probing the functional heterogeneity of surface binding sites by analysis of experimental binding traces and the effect of mass transport limitation. Biophys.J. (92): 1742-1758; 2007.
Let us know what your results are.
Arnoud
Please Log in or Create an account to join the conversation.